- 品牌
- 司鼎;OriCell
聚合酶链式反应:反应的控制:PCR反应的缓冲液 提供合适的酸碱度与某些离子;镁离子浓度 总量应比dNTPs的浓度高,常用1.5mmol/L;底物浓度 dNTP以等摩尔浓度配制,20~200umol/L;TaqDNA聚合酶 2.5U(100ul);引物浓度一般为0.1 ~ 0.5umol/L;反应温度和循环次数;变性温度和时间 95℃,30s;退火温度和时间 低于引物Tm值5 ℃左右,一般在45~55℃;延伸温度和时间 72℃,1min/kb(10kb内);Tm值=4(G+C) +2(A+T);循环次数 :一般为25 ~ 30次。循环数决定PCR扩增的产量。模板初始浓度低,可增加循环数以便达到有效的 扩增量。但循环数并不是可以无限增加的。一般循环数为30个左右,循环数超过30个以后,DNA聚合酶活性逐渐达到饱和,产物的量不再随循环数的增加而增加,出现了所谓的“平台期”。在嵌套聚合酶链反应中,除了预期的靶之外,该产物可能仍然由非特异性扩增的DN段组成。莆田骨头数字PCR

绝大多数聚合酶链反应方法依赖于热循环。热循环将反应物暴露于加热和冷却的重复循环中,以允许不同的温度依赖性反应——具体地说,脱氧核糖核酸融化和酶-驱动DNA复制。聚合酶链反应使用两种主要试剂-引物(引物是短的单链DN段,称为寡核苷酸,是目标DNA区域的互补序列)和DNA聚合酶。在PCR反应的步,DNA双螺旋结构的两条链在高温下物理分离,这个过程称为脱氧核糖核酸变性。第二步,降低温度,引物与互补的脱氧核糖核酸序列结合。这两条DNA链就变成了模板,以酶促的方式从构成DNA的自由核苷酸中组装出一条新的DNA链。随着聚合酶链反应的进行,产生的DNA本身被用作复制的模板,启动了一个连锁反应,原始的DNA模板是以指数形式放大。南京实时荧光定量PCR设计公司聚合酶链反应具有定量合成产物的优点。

聚合酶链式反应:Taq(水生栖热菌)聚合酶耐热DNA聚合酶的很好活性温度约为75-80°C(167-176°F),尽管该酶通常使用72°C(162°F)的温度。在该步骤中,DNA聚合酶通过从反应混合物中添加在5’-至-3’方向上与模板互补的游离DNA链来合成与DNA模板链互补的新DNA链,在新生(伸长)DNA链的末端,将dNTPs的5'-磷酸基与3'-羟基缩合。延伸所需的精确时间取决于所使用的DNA聚合酶和要扩增的DNA目标区域的长度。根据经验,在很好温度下,大多数DNA聚合酶每分钟聚合一千个碱基。在很好条件下(即,如果没有限制底物或试剂的限制),在每个延伸/延伸步骤中,DNA靶序列的数量加倍。随着每一个连续的循环,原始模板链加上所有新产生的链成为下一轮延伸的模板链,导致特定DNA目标区域的指数(几何)扩增。
聚合酶链反应:热启动聚合酶链式反应:一种在PCR的初始建立阶段减少非特异性扩增的技术。它可以通过将反应组分加热到变性温度(例如95 c)在加入聚合酶。[36]已经开发了特殊的酶系统,通过结合抗体来抑制聚合酶在环境温度下的活性[37][37]或者通过共价键结合的抑制剂的存在,该抑制剂在高温活化步骤后解离。热启动/冷完成聚合酶链反应是通过新的杂合聚合酶实现的,这些酶在环境温度下不活跃,在伸长温度下立即。序列间特异性聚合酶链反应(ISSR):一种用于DNA指纹识别的聚合酶链反应方法,它放大简单重复序列之间的区域,以产生扩增片段长度的独特指纹。电子聚合酶链反应(数字聚合酶链反应、虚拟聚合酶链反应、电子聚合酶链反应、电子聚合酶链反应)是指计算工具使用给定的一组引物 ( 探针)来扩增来自测序的基因组或转录组的DNA 序列,用于计算理论聚合酶链反应结果。电子PCR被提出作为分子生物学的教育工具。聚合酶链反应可以选择这些突变来理解蛋白质是如何完成其功能的,并改变或改善蛋白质功能。
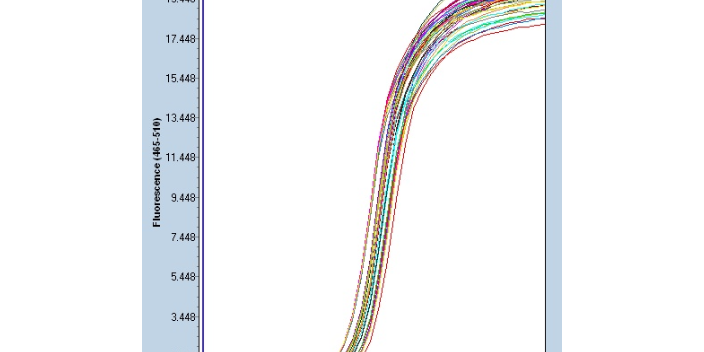
聚合酶链反应的一个主要限制是,为了产生允许其选择性扩增的引物,需要关于目标序列的先前信息。这意味着,通常情况下,PCR用户必须知道两个单链模板中每个模板上目标区域上游的精确序列,以确保DNA聚合酶正确结合引物-模板杂交体,并随后在DNA合成过程中产生整个目标区域。像所有酶一样,DNA聚合酶也容易出错,这反过来会导致产生的PCR片段发生突变。PCR的另一个限制是,即使是很少量的污染DNA也可以被扩增,导致误导或模糊的结果。为了很大限度地减少污染的可能性,调查人员应该为试剂制备、聚合酶链反应和产品分析预留单独的房间。试剂应分配到一次性的等分试样中。应经常使用带有一次性柱塞和超长移液器吸头的移液器。逆转录聚合酶链反应(逆转录-聚合酶链反应):用于从RNA中扩增DNA。南京实时荧光定量PCR设计公司
PCR反应的特异性决定因素为:引物与模板DNA特异正确的结合。莆田骨头数字PCR
聚合酶链反应的常见问题分析与解决方法:反应缓冲液未完全融化或未充分混匀。确保反应缓冲液融化完全并彻底混匀。引物特异性差。利用BLAST检查引物特异性或重新设计引物。引物量过多。减少反应体系中引物的用量。模板量过多。质粒DNA的用量应<50ng,而基因组DNA则应<200ng。外源DNA污染。确保操作的洁净。阴性对照出现条带:试剂,头,工作台污染。使用全新的试剂和头,对工作台进行清洁。条带大小与理论不符:污染。使用全新的试剂和头,对工作台进行清洁。模板或引物使用错误。更换引物和模板。基因亚型。对研究的基因进行序列分析和BLAST研究。莆田骨头数字PCR
上海司鼎生物科技有限公司是一家有着雄厚实力背景、信誉可靠、励精图治、展望未来、有梦想有目标,有组织有体系的公司,坚持于带领员工在未来的道路上大放光明,携手共画蓝图,在上海市等地区的医药健康行业中积累了大批忠诚的客户粉丝源,也收获了良好的用户口碑,为公司的发展奠定的良好的行业基础,也希望未来公司能成为行业的翘楚,努力为行业领域的发展奉献出自己的一份力量,我们相信精益求精的工作态度和不断的完善创新理念以及自强不息,斗志昂扬的的企业精神将引领上海司鼎生物科技供应和您一起携手步入辉煌,共创佳绩,一直以来,公司贯彻执行科学管理、创新发展、诚实守信的方针,员工精诚努力,协同奋取,以品质、服务来赢得市场,我们一直在路上!

Real-time PCR:所谓Real-time PCR技术,是指在PCR反应体系中加入萤光基团,利用萤光信号累积实时监测整个PCR进程,之后通过标準曲线对未知模板进行定量分析的方法。利用萤光信号的变化实时检测PCR扩增反应中每一个循环扩增产物量的变化,通过Ct值和标準曲线的分析对起始模板进行定量分析。Real-time PCR技术即实时萤光定量PCR避免了传统PCR以终产物监测定量产生的偏差,提高实验的重複性。该技术已经被普遍用于监测细胞mRNA表达量的变化;比较不同组织的mRNA表达差异;验证基因晶片,siRNA干扰的实验结果。Real-time PCR在实验过程中要防止RNA的降解。无...
- 武汉特殊样本Real-time PCR研究方案 2024-12-26
- 珠海特殊样本定量PCR技术服务 2024-12-26
- 深圳骨头荧光PCR方案 2024-12-26
- 南京特殊样本RT-PCR检测技术 2024-12-25
- 嘉兴微量Real-time PCR 2024-12-25
- 温州血液荧光定量PCR服务 2023-08-12
- 深圳分子生物学RT-PCR检测技术方案 2023-08-11
- 无锡分子生物学RT-PCR检测技术方案 2023-08-11
- 莆田骨头定量PCR哪家好 2023-08-11
- 扬州骨头荧光定量PCR 2023-08-11


- 常州微量数字PCR技术服务 2023-08-10
- 上海特殊样本荧光定量PCR服务 2023-08-09
- 珠海特殊样本PCR检测技术供应商 2023-08-09
- 温州实时定量PCR原理 2023-08-09
- 杭州特殊样本Real-time PCR原理及步骤 2023-08-09
- 温州骨头数字PCR设计公司 2023-08-08

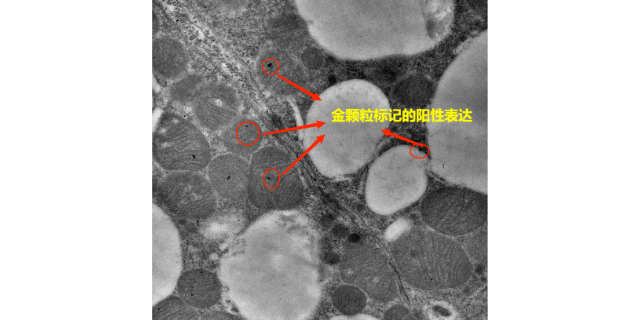
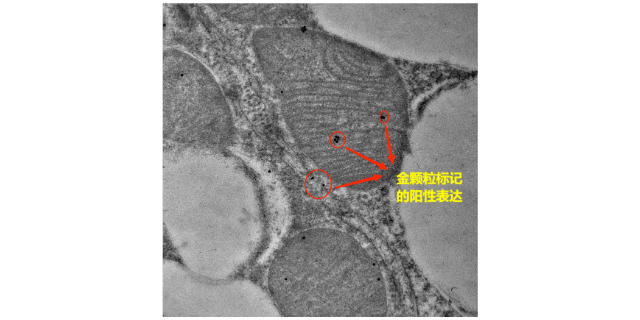
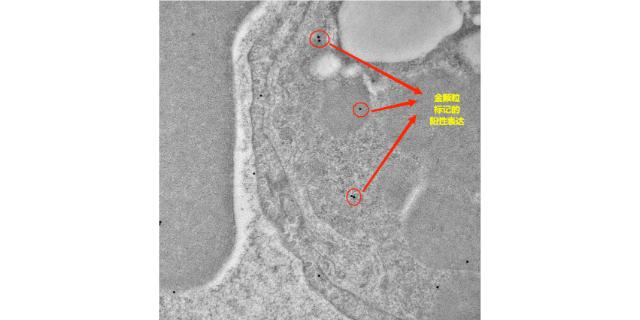
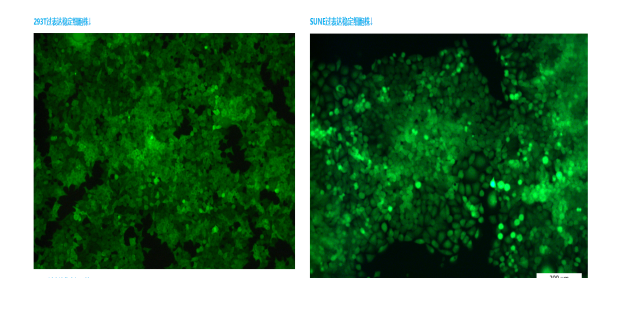
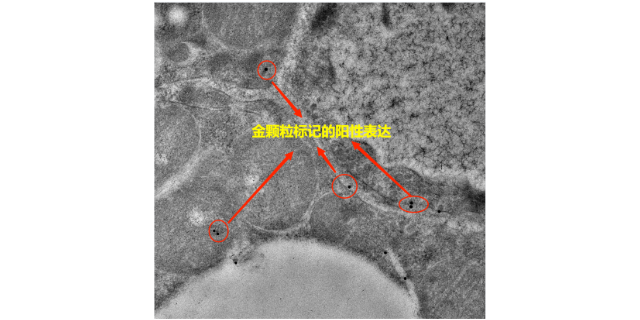
- 徐州超微结构免疫电镜检测 01-02
- 黄石细胞增殖与毒性检测服务特点 01-02
- 徐州高效多种细胞培养及检测服务哪里有 01-02
- 厦门发病机理免疫电镜检测原理 01-02
- 襄阳抗原定位免疫电镜技术方案 01-02
- 蚌埠超微结构免疫电镜检测原理 01-01
- 黄石发病机理免疫电镜技术特点 01-01
- 蚌埠抗体反应免疫电镜技术哪家靠谱 01-01
- 无锡抗原定位免疫电镜技术哪家好 01-01
- 东莞抗原定位免疫电镜技术方案 01-01